sphinx-quickstart on Wed Jan 29 13:25:56 2020. You can adapt this file completely to your liking, but it should at least contain the root toctree directive.
JBrowse中文翻译¶
注解
本人在开发时阅读的JBrowse文档,仅供参考学习
JBrowse中文文档¶
安装¶
一个最通常的情况是,下载JBrowse之后,将整个项目文件夹放在web服务器的目录下(在ubuntu中通常是/var/www/),JBrowse就是一个静态站点, 它通过index.html中的js进行数据处理,不需要后端处理.
安装JBrowse之前需要做什么¶
有一些前提条件可以帮助您进行JBrowse设置,包括
- unix操作系统系列-- MacOSX, Linux, 或者 WSL on Windows(在windows10的应用商店中下载ubuntu,使用cmd输入命令
bash即可进入linux环境) - Web服务器-JBrowse是一组静态文件,可与Apache或Nginx一起使用
- 命令行技能-熟悉命令行将帮助您更好的使用本教程
- Sudo访问-sudo是没有必要的,除非您需要它来修改Web服务器文件,例如 在/ var / www中,修改权限
如果您不具备所有这些条件,请考虑使用JBrowse Desktop,因为它不需要命令行,并且易于在所有操作系统上使用:)
导入JBrowse插件¶
如果您使用的是JBrowse插件,则还需要安装Node.js版本6或更高版本。请按照https://nodejs.org/en/download/package-manager/#debian-and-ubuntu-based-linux-distributions 中的步骤在ubuntu上安装node.js
下载JBrowse的发行版本¶
curl -L -O https://github.com/GMOD/jbrowse/releases/download/1.16.7-release/JBrowse-1.16.7.zip
unzip JBrowse-1.16.7.zip
sudo mv JBrowse-1.16.7 /var/www/html/jbrowse
cd /var/www/html
sudo chown `whoami` jbrowse
cd jbrowse
./setup.sh
注解
文档假设你已经安装了web服务器,例如apache.
在ubuntu上安装apache使用命令 apt install apache2 会在/var中自动生成www文件夹,里面会有index.html初始文件.
访问ubuntu服务器ip即可查看该页面.将JBrowse项目文件夹放入/var/www中,配置apache,apache具体配置自行搜索
如果将JBrowse作为插件需要备用JBrowse设置¶
JBrowse现在在构建时捆绑了插件,因此,如果您使用插件或自己修改jbrowse源代码,则必须下载源代码https://github.com/GMOD/jbrowse
git clone https://github.com/gmod/jbrowse
cd jbrowse
git checkout 1.16.7-release # or version of your choice
./setup.sh
npm run start # starts a express.js dev server on port 8082, alternatively move the entire jbrowse dir to /var/www/html as above
注解
使用npm run watch自动获取对您所做的代码所做的更改(但是,如果添加或删除文件,则需要重新启动) 对于中国的用户,如果使用自定义配置,推荐使用npm镜像
npm config set registry http://r.cnpmjs.org
npm config set puppeteer_download_host=http://cnpmjs.org/mirrors
export ELECTRON_MIRROR="http://cnpmjs.org/mirrors/electron/"
祝贺!¶
您应该看到一条消息,“Congratulations, JBrowse is on the web”,并带有指向“ Volvox example data”的链接。 如果您没有看到此消息,则可能是错过了设置步骤
索引文件格式¶
在本节中,我们将使用“索引文件格式”,以纯文本配置为例
加载FASTA格式的索引¶
首先,我们假设正在建立Volvox mythicus(Volvox属中的一个神话物种)的基因组。 Volvox基因组已由测序中心在2018年进行了测序,他们想立即设置JBrowse。 他们为我们提供了指向其FASTA文件的链接,我们将下载该文件
mkdir data
curl -L https://jbrowse.org/code/JBrowse-1.16.7/docs/tutorial/data_files/volvox.fa > data/volvox.fa
我们将使用samtools的 faidx 命令来创建“ FASTA index”.
FASTA index能将非常大的FASTA文件“按需”下载到JBrowse中.
例如, 仅下载特定视图所需的序列.
samtools faidx data/volvox.fa
FASTA index将是一个名为volvox.fa.fai的文件。 然后,我们将这些文件移到JBrowse可以使用的“数据目录”中
然后创建文件data / tracks.conf,使用以下内容
[GENERAL]
refSeqs=volvox.fa.fai
[tracks.refseq]
urlTemplate=volvox.fa
storeClass=JBrowse/Store/SeqFeature/IndexedFasta
type=Sequence
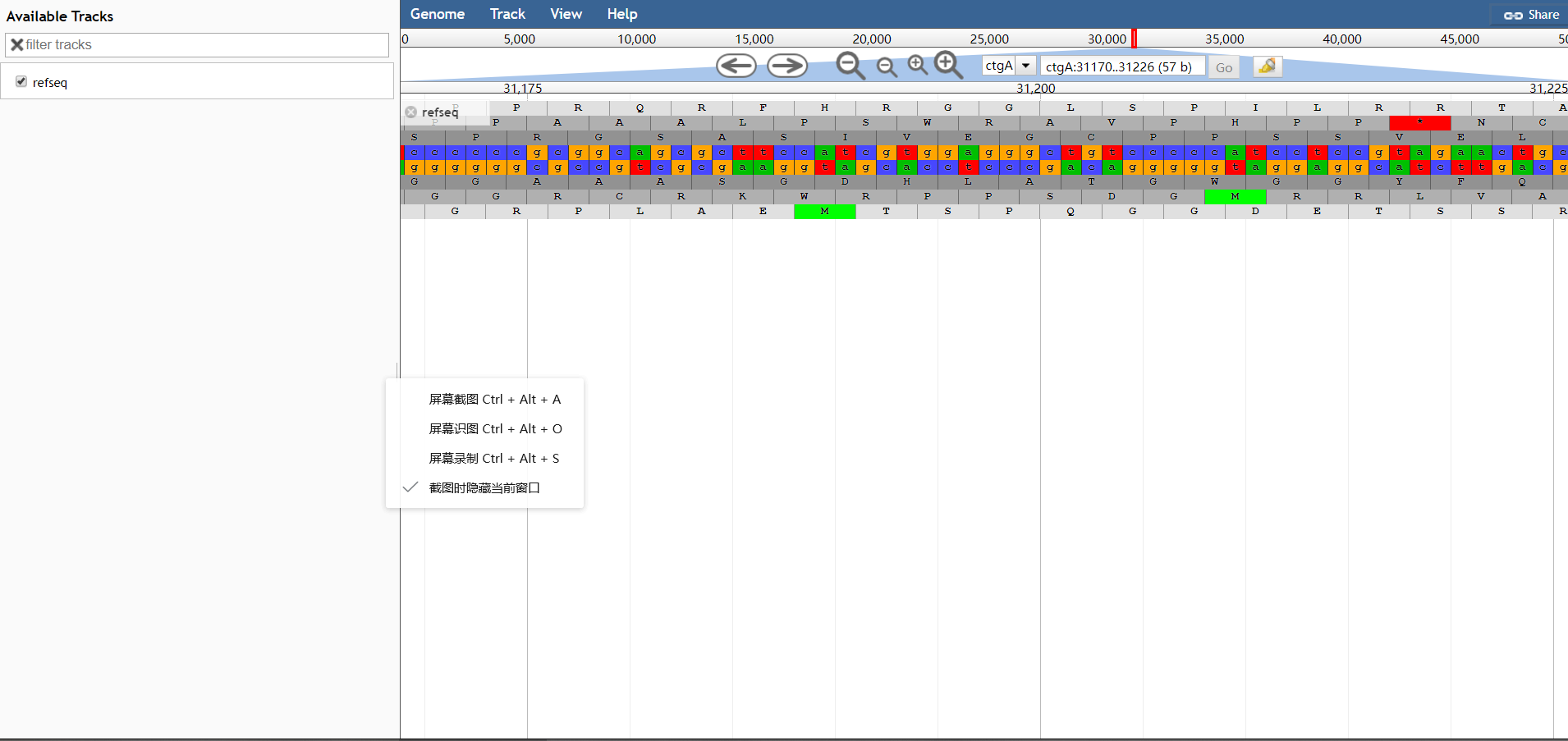
此时,您应该可以打开http://localhost/jbrowse/?data=data(或者打开http://localhost/jbrowse/), 您就会看到带有参考序列轨迹的基因组。 示例图如下:
加载Tabix GFF3¶
我们将使用的新生成的“基因注释”文件
curl -L https://jbrowse.org/code/JBrowse-1.16.7/docs/tutorial/data_files/volvox.gff3 > data/volvox.gff3
当我们处理GFF3以在JBrowse中使用时,我们的目标是使用GFF3Tabix格式。 T abix格式允许随机访问类似于Indexed FASTA的基因组区域。因此, 我们必须首先对GFF进行排序以为Tabx做准备
sort -k1,1 -k4,4n data/volvox.gff3 > data/volvox.sorted.gff3
bgzip data/volvox.sorted.gff3
tabix -p gff data/volvox.sorted.gff3.gz
在data/tracks.conf中加入一下配置:
[tracks.genes]
urlTemplate=volvox.sorted.gff3.gz
storeClass=JBrowse/Store/SeqFeature/GFF3Tabix
type=CanvasFeatures
完成!
加载BAM¶
如果已获得序列比对,也可以创建一个显示比对的“比对”轨迹 对于volvox,我们得到一个文件
curl -L https://jbrowse.org/code/JBrowse-1.16.7/docs/tutorial/data_files/volvox-sorted.bam > data/volvox-sorted.bam
请注意,此BAM文件已经排序。 如果您的BAM未排序,则必须对其进行排序以在JBrowse中使用。使用以下命令行:
samtools index data/volvox-sorted.bam
最后在data/tracks.conf中添加以下内容:
[tracks.alignments]
urlTemplate=volvox-sorted.bam
storeClass=JBrowse/Store/SeqFeature/BAM
type=Alignments2
检查您的文件是否已经加载成功¶
此时,如果jbrowse文件位于您的Web服务器中,则应具有目录布局,例如
/var/www/html/jbrowse
/var/www/html/jbrowse/data
/var/www/html/jbrowse/data/volvox.fa
/var/www/html/jbrowse/data/volvox.fa.fai
/var/www/html/jbrowse/data/volvox.sorted.gff3.gz
/var/www/html/jbrowse/data/volvox.sorted.gff3.gz.tbi
/var/www/html/jbrowse/data/volvox-sorted.bam
/var/www/html/jbrowse/data/volvox-sorted.bam.bai
/var/www/html/jbrowse/data/tracks.conf
你的tracks.conf文件夹中的内容如下:
[GENERAL]
refSeqs=volvox.fa.fai
[tracks.refseq]
urlTemplate=volvox.fa
storeClass=JBrowse/Store/SeqFeature/IndexedFasta
type=Sequence
[tracks.genes]
urlTemplate=volvox.sorted.gff3.gz
storeClass=JBrowse/Store/SeqFeature/GFF3Tabix
type=CanvasFeatures
[tracks.alignments]
urlTemplate=volvox-sorted.bam
storeClass=JBrowse/Store/SeqFeature/BAM
type=Alignments2
然后,您可以访问http:// localhost / jbrowse /将自动加载“data”目录。
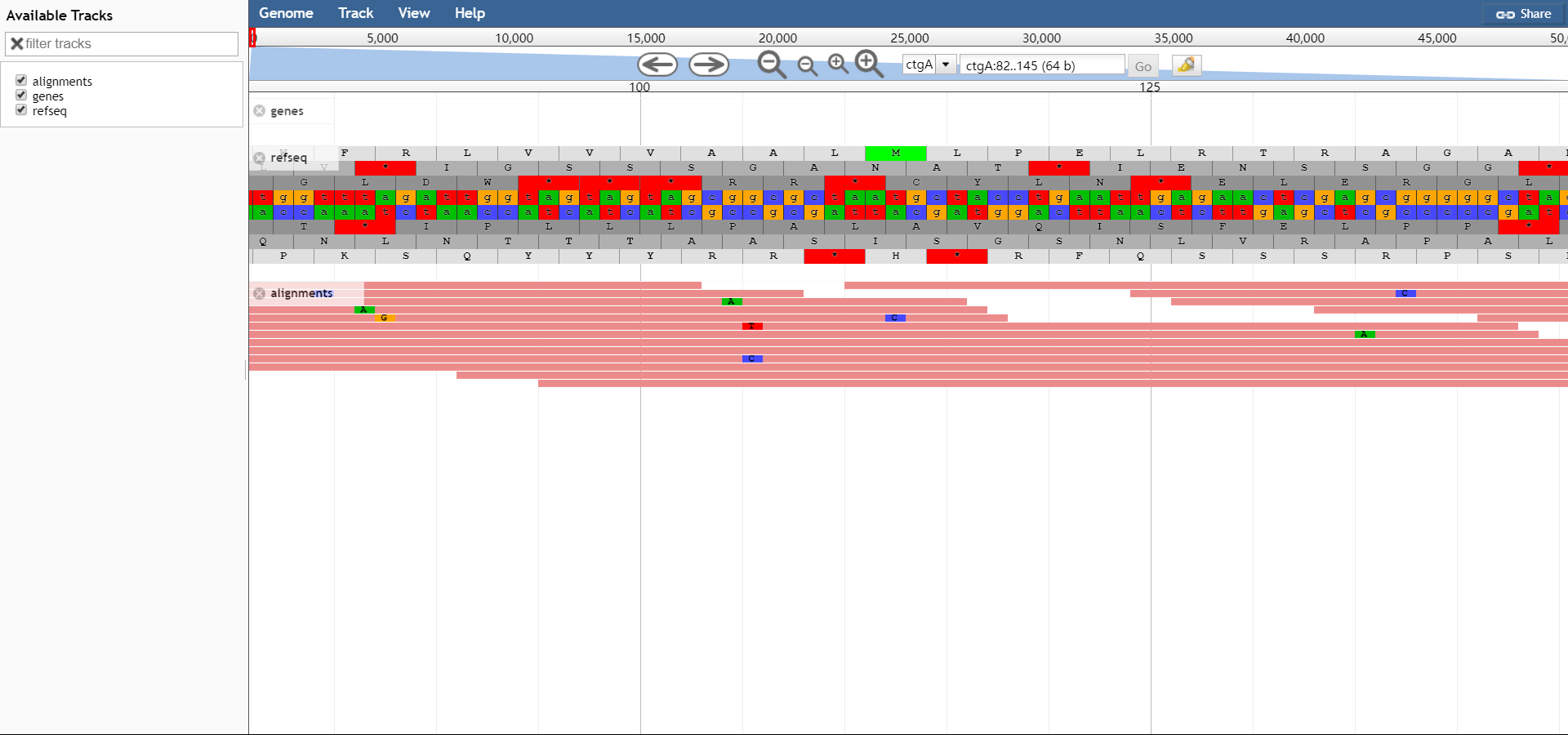
示例图如下:
祝贺!¶
您现在成功配置了JBrowse.
经典快速入门指南¶
参考序列¶
在加载注释数据之前,请使用 bin/prepare-refseqs.pl 格式化JBrowse的参考序列。
对于FASTA格式序列文件
bin/prepare-refseqs.pl --fasta docs/tutorial/data_files/volvox.fa
对于已经存储在Bio :: DB :: *数据库中的序列,可以将prepare-refseqs.pl与biodb-to-json.pl配置结合使用
bin/prepare-refseqs.pl --conf docs/tutorial/conf_files/volvox.json
基因组注释和特征¶
JBrowse可以从平面文件或具有Bio :: DB :: * Perl接口的数据库中导入数据。
bin/flatfile-to-json.pl¶
如果您有平面文件,例如GFF3或BED,通常最好使用bin / flatfile-to-json.pl导入它们。 bin / flatfile-to-json.pl接受许多不同的命令行设置, 这些设置可用于自定义新track的外观。 运行bin / flatfile-to-json.pl --help查看可用设置的描述。
bin/flatfile-to-json.pl --gff path/to/my.gff3 --trackType CanvasFeatures --trackLabel mygff
bin/biodb-to-json.pl¶
如果您拥有基因组注释数据库,例如Chado,Bio :: DB :: SeqFeature :: Store或Bio :: DB :: GFF,则可以使用JBrowse的biodb-to-json.pl。 您将bin / biodb-to-json.pl与配置文件一起使用(其格式在此处记录)。
bin/biodb-to-json.pl --conf docs/tutorial/conf_files/volvox.json
Next-gen reads (BAM)¶
JBrowse可以直接从BAM文件显示对齐方式,而无需进行预处理。 要使用该文件,请将BAM文件和BAM索引下载到您的数据文件夹中, 然后您可以将文本片段手动编辑到tracks.conf中:
[tracks.alignments]
storeClass = JBrowse/Store/SeqFeature/BAM
urlTemplate = myfile.bam
category = NGS
type = JBrowse/View/Track/Alignments2
key = BAM alignments from SEQ1654
然后,你需要在data文件夹中存在以下文件:
data/myfile.bam
data/myfile.bam.bai
data/tracks.conf
在tracks.conf包含上述配置的地方,然后myfile.bam将相对于tracks.conf的位置进行定位, 因此由于它们位于同一目录中,因此不需要任何其他路径限定。 请注意,urlTemplate可以是完整的URL路径.
另请注意:BAM文件需要排序和索引(即具有对应的.bai文件或.csi文件。如果为.csi,则可以使用csiUrlTemplate。 如果为.bai,则将自动定位它 作为<yourbamfile.bam>,最后加上.bai)。
Next-gen read track类型¶
JBrowse有两种主要的轨道类型,这些轨道类型专门设计用于BAM数据:
Alignments2 显示BAM文件中的各个比对,以及BAM的MD或CIGAR字段中编码的插入,删除,跳过的区域和SNP。
SNPCoverage 显示覆盖度直方图,彩色条显示碱基水平不匹配和读数中可能的SNP的位置。
要使用它们,请在轨道配置中设置type = Alignments2或type = SNPCoverage。
Index Names¶
加载要素数据后,要让用户通过在自动完成搜索框中键入要素名称或ID来查找要素,必须使用 bin/generate-names.pl 生成要素名称的特殊索引。
bin/generate-names.pl -v
注意:每次使用任何* -to-json.pl脚本向JBrowse添加新注释时,都需要重新运行 bin/generate-names.pl 将新功能名称添加到索引。
还要注意:bin / generate-names.pl索引的轨道类型包括:
- GFF,GBK,BED通过flatfile-to-json.pl加载。 默认情况下,对“ ID”,“名称”和“别名”建立索引。 请注意,--nameAttributes可用于索引其他字段
- Features from biodb-to-json.pl
- VCF tabix文件和GFF3 tabix文件(对它们从GFF的“ ID”和“名称”字段建立索引,并且仅对来自VCF的ID进行索引)
BAM的reads和BigWigs不会由generate-names.pl编制索引
定量轨道(BigWig和Wiggle)¶
JBrowse可以直接从BigWig文件显示对齐方式,而无需进行预处理。 只需将带有文件相对URL的节添加到您的data / tracks.conf文件中,格式为:
[ tracks.my-bigwig-track ]
storeClass = JBrowse/Store/SeqFeature/BigWig
urlTemplate = myfile.bw
type = JBrowse/View/Track/Wiggle/XYPlot
key = Coverage plot of NGS alignments from XYZ
JBrowse有两种专门用于定量数据的track类型:
- JBrowse/View/Track/Wiggle/XYPlot
将定量数据显示为条形图。 有关配置选项,请参见JBrowse Wiki。
- JBrowse/View/Track/Wiggle/Density
将定量数据显示为“热图”图,默认情况下,其具有正分数的区域绘制为逐渐变深的蓝色,而具有负分数的区域绘制为逐渐变深的红色。 有关配置选项,请参见JBrowse Wiki,包括如何更改颜色改变点(bicolor_pivot)和颜色。
要使用它们,请在轨道配置中设置type = JBrowse / View / Track / Wiggle / XYPlot或type = JBrowse / View / Track / Wiggle / Density。
结论¶
祝您好运,并享受JBrowse!